- 移动端


MedChemExpress LLC品牌商
14 年
手机商铺
- NaN
- 0
- 0
- 2
- 2

推荐产品




公司新闻/正文
药物研发:抗菌药物药效(PD)药代(PK)的给药逻辑
286 人阅读发布时间:2026-06-07 09:59
Section.01
药物代谢动力学 (PK)
药物代谢动力学 (Pharmacokinetics, PK) :简称药代动力学,是定量研究药物在生物体内吸收 (Absorption) 、分布 (Distribution) 、代谢 (Metabolism) 和排泄 (Excretion) ,随时间变化规律的学科。
——简单理解,PK 探究:在药物摄入后,机体是如何处理药物的?核心是测定药物浓度随时间的变化。
知识链接
1. 药物的体内动力学过程
• 动力学本身分为零级动力学 (定值) 和一级动力学 (线性动力学) 。
• 目前临床上应用的大多数小分子药物,其体内的吸收、分布、代谢、排泄 (ADME) 过程都遵循一级动力学特征 (线性动力学特征) 。
2. 给药方式
• 血管内给药:如静脉注射、静脉滴注
• 血管外给药:如口服、肌内注射、吸入给药、透皮给药
如何探究?药代动力学通过非房室模型或房室模型进行建模分析。房室模型 (Compartment Model) 使用动力学模型估算血药浓度-时间图。被更广泛地使用的是——非房室模型 (Noncompartmental Analysis, NCA) ,通过估算血药浓度-时间图中的曲线下的面积,来估计药物暴露量。
血药浓度-时间曲线下面积 (AUC)
首先,了解下血药浓度,是指药物吸收后在血浆内的总浓度,这包括了与血浆蛋白结合的药物或在血浆中游离的药物,有时也可泛指药物在全血中的浓度。
血药浓度-时间曲线下面积 (Area under curve,AUC) 指药物在血液中的浓度随时间变化曲线 (药-时曲线) 和横坐标所包围的面积,其数值反映在某段时间内进入人体循环的药量,即药物的吸收程度。
——评价药物吸收程度的重要指标。
-
药物暴露量:直接反映药物在体内被吸收和暴露的总量。AUC 越大,代表药物在体内停留的时间越长、浓度越高。
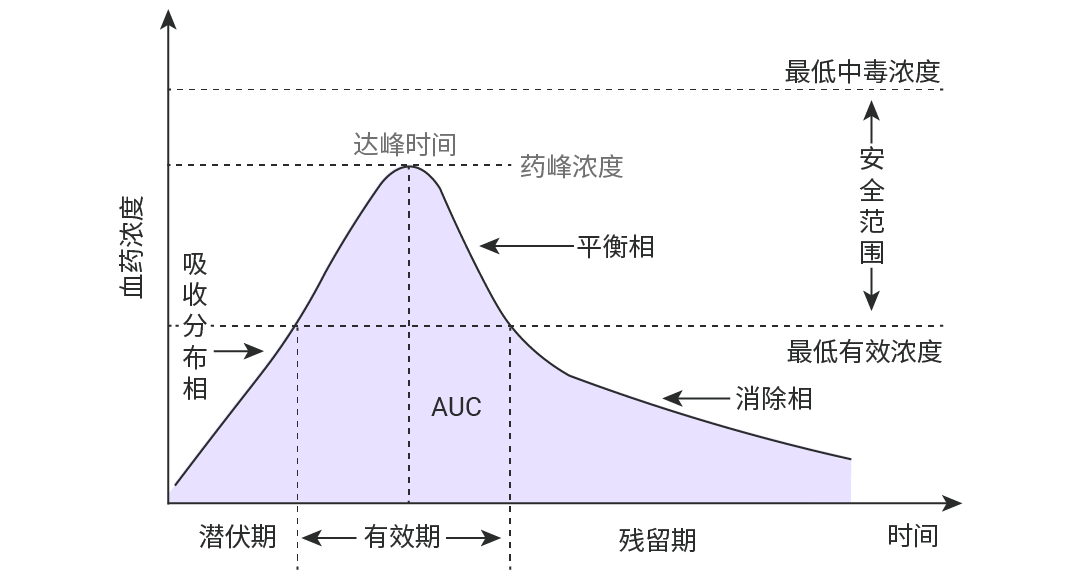
该曲线以时间为横坐标,血药浓度为纵坐标。
AUC0→t表示从给药开始到t时刻的药-时曲线下面积;
AUC0→∞则指从给药开始到所有原形药物全部消除为止时的曲线下总面积。
可通过梯形法则 (数值积分) 计算各单位间隔时间内面积之和 (最常用的手段) 。取样时间点 (采样点) 越密集,预测的梯形面积越能反映浓度-时间曲线的实际情况。
采样点
采样点的确定对药代动力学研究结果有重大影响,若采样点过少或选择不当,得到的血药浓度-时间曲线可能与药物在体内的真实情况产生较大差异。
① 给药前需要采血作为空白样品。
② 为获得给药后的一个完整的血药浓度-时间曲线,采样时间点的设计应兼顾药物的吸收相、平衡相 (峰浓度附近) 和消除相。
③ 一般在吸收相至少需要 2~3 个采样点,对于吸收快的血管外给药的药物,应尽量避免第一个点是峰浓度 (Cmax);在 Cmax 附近至少需要 3 个采样点;消除相需要 4-6 个采样点。
④ 整个采样时间至少应持续到 3-5 个半衰期,或持续到血药浓度为 Cmax 的 1/10-1/20。
⑤ 为保证最佳采样点,建议在正式试验前选择 2~3 只动物进行预试验,然后根据预试验的结果,审核并修正原设计的采样点。
根据试验中测得的各受试动物的血药浓度-时间数据,可求得受试药物的主要药代动力学参数。
例如:静脉注射给药,应提供 t1/2、Vd、 AUC、Cl 等参数值;血管外给药,除提供上述参数外,还应提供 Cmax 和 Tmax 等参数,以反映药物吸收的规律。另外,提供统计矩参数,如: MRT、AUC0→t 和 AUC0→∞ 等,对于描述药物药代动力学特征也是有意义的[1]。
Cmax (药峰浓度) 和 Tmax (药物达峰时间)
药峰浓度,代表符号是 Cmax,是指血管外给药吸收后所能达到的最高血浆药物浓度,即血药浓度—时间曲线 (药-时曲线) 上的最大血药浓度值。单位以 μg/mL 或 mg/L 表示。当 Cmax 达到一定浓度药物才能显效,浓度越高效果越强,但若超出了安全范围则可显示出毒性反应。
药物达峰时间,代表符号是 Tmax,是指机体在给药后,血液中的药物浓度从零开始上升,直至达到最高点 Cmax 时所需的时间。Tmax 是反映药物被身体吸收快慢的重要指标,被作为药物起效的标志。Tmax 数值越小,起效越快,可被用于指导用药间隔,是判断药物起效时间和设计给药频次的重要依据。
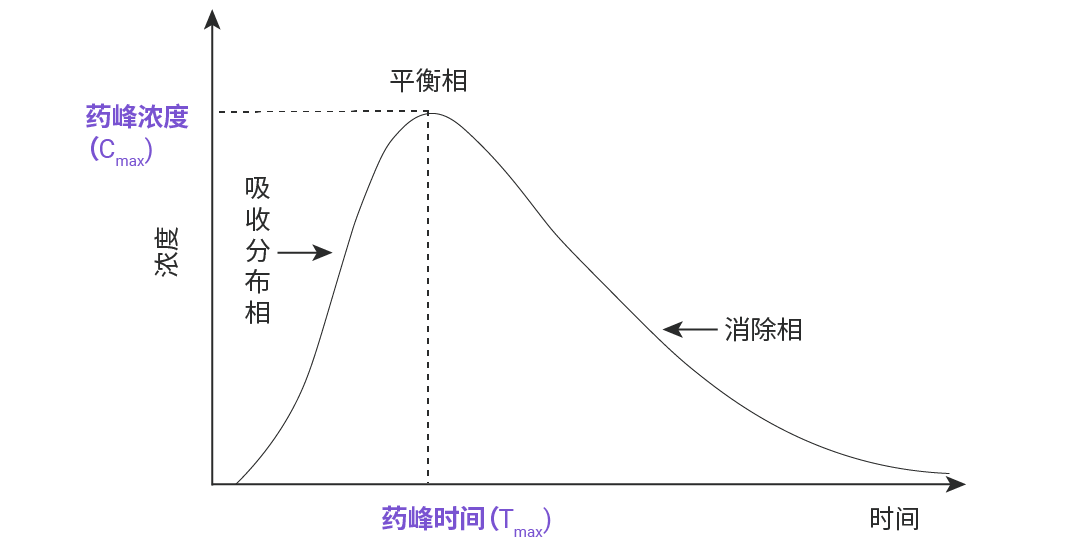
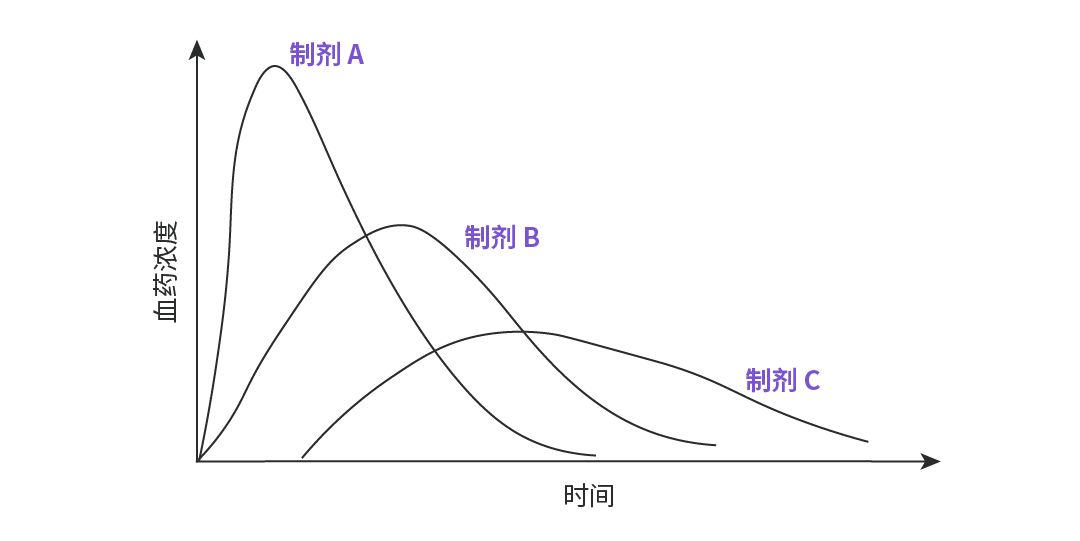
——反映药物在体内吸收速率的两个重要指标,药物的吸收速度快,则其峰浓度高,达峰时间短。如图 3 所示,图中 A、B、C 三个制剂的吸收程度相似,但吸收速度不同,其中吸收速度 A>B>C。
t1/2 (半衰期)
清除半衰期,代表符号是 t1/2,是指血药药物浓度下降一半所需的时间。与消除速率常数 (elimination rate constant, k) 存在倒数关系,公式为:t1/2=0.693/k 。
由于 t1/2更直观,所以临床上多用 t1/2 来反映药物消除的快慢,它是临床确定给药间隔长短的重要依据之一。一般情况下,经过 5-6 个 t1/2 达到稳态血药浓度,药物可基本消除干净。
——t1/2 反映药物消除快慢的程度及机体消除药物的能力。
-
特征参数:不因药物剂型、给药途径或剂量而改变。
Vd (表观分布容积)
表观分布容积,代表符号是 Vd,是指药物在体内按血中同样浓度分布所需体液的总容积,单位是 L 或 L/kg。
Vd 值的大小反映药物在体内分布的广泛浓度以及组织结合程度,Vd 值并不代表真正的生理体积,其意义在于反映药物的体内分布范围。
Vd 值在不同的药代动力学模型中不同:
-
非房室模型 (NCA) 中,Vd 通常会细分为两种最常用的计算指标:消除相分布容积 (Vz) 和稳态分布容积 (Vss) 。
-
对于单室模型的药物而言,分布容积与体内药量 X 和血药浓度 C 之间存在下列关系: Vd=X/C。
药物的分布容积的大小取决于其脂溶性、膜通透性、组织分配系数及药物与血浆蛋白等生物物质的结合率等因素。如药物的血浆蛋白结合率高,则其组织分布较少,血药浓度高。
我们可以根据体液的分布情况 (见表 1),由药物的分布容积可以粗略地推测其在体内的大致分布情况。
表 1. 体液的分布情况。

【以单室模型为例】
① 若一个药物的 Vd 为 3-5 L 左右,那么这个药物可能主要分布于血液并与血浆蛋白大量结合,如双香豆素、苯妥英钠和保泰松等;
② 若一个药物的 Vd 为 10-20 L 左右,则说明这个药物主要分布于血浆和细胞外液,这类药物往往不易通过细胞膜,因此无法进入细胞内液,如溴化物和碘化物等;
③ 若一个药物的分布容积为 40 L,则这个药物可以分布于血浆和细胞内、外液,表明其在体内的分布较广,如安替比林;
④ 有些药物的 Vd 非常大,可以达到 100 L 以上,这一体积已远远地超过了体液的总容积,这类药物在体内往往有特异性的组织分布,如硫喷妥钠具有较高的脂溶性,可以大量地分布于脂肪组织,而 I131 可以大量地浓集于甲状腺,因而其分布容积也很大。
——由此可见我们可以通过分布容积来了解药物在体内的分布情况。
Cl (清除率)
清除率,代表符号是 Cl,是指单位时间内,机体能将多少容积含药血浆从体内清除,单位是 L/h 或 L/h/kg,表示从血中清除药物的速率或效率。
——反映药物从体内消除的一个重要参数。
清除率与清除速率常数k和分布容积之间的关系可用公式:Cl=k · vd 表示。
F (生物利用度)
生物利用度,代表符号是 F,是指药物被机体吸收进入全身血液循环的速度和程度。简单来说,就是摄入的药中,实际有多少比例真正进入血液并发挥了药效。通常来讲,静脉注射的 F 为 100%,起效最快、完全吸收。
——评价药物吸收程度的重要指标。
生物利用度可以分为绝对生物利用度和相对生物利用度。同一种药物,不同的制剂,生物利用度是不同的。同一制剂,不同厂家产品的生物利用度往往也是不同的,甚至同一厂家的制剂,不同的生产批,次也可能出现生物利用度的差异,从而影响药物疗效和安全性。
-
绝对生物利用度:药物进入体循环的剂量占给药总剂量的分数。侧重评估静脉给药或血管外给药两种给药途径的吸收差异。F=AUC 血管外给药/AUC 静脉给药 × 100%
-
相对生物利用度:同一血管外给药途径的某一种药物制剂(如不同剂型、不同药厂)的 AUC 与已知的参比制剂进行比较。侧重评估给药不同制剂类型或厂家的吸收差异。F=AUC 受试试剂/AUC 标准制剂×100%
为什么有些受试药 F 值很低?
药物从口服到进入血液,主要会经过以下损耗:
(1) 首过效应:口服药物被吸收后,会先经过肠壁和肝脏,部分药物在到达全身循环前就被代谢掉了。
(2) 吸收不完全:药物在胃肠道不稳定 (被胃酸破坏) ,或因分子太大无法穿过肠壁。
Section.02
药物效应动力学 (PD)
药物进入机体后,出现两种不同的效应。除了上述提到的药代动力学,还包括药物对机体产生的治疗作用和毒副作用,即所谓的药效学和毒理学,也就是药物如何影响机体?
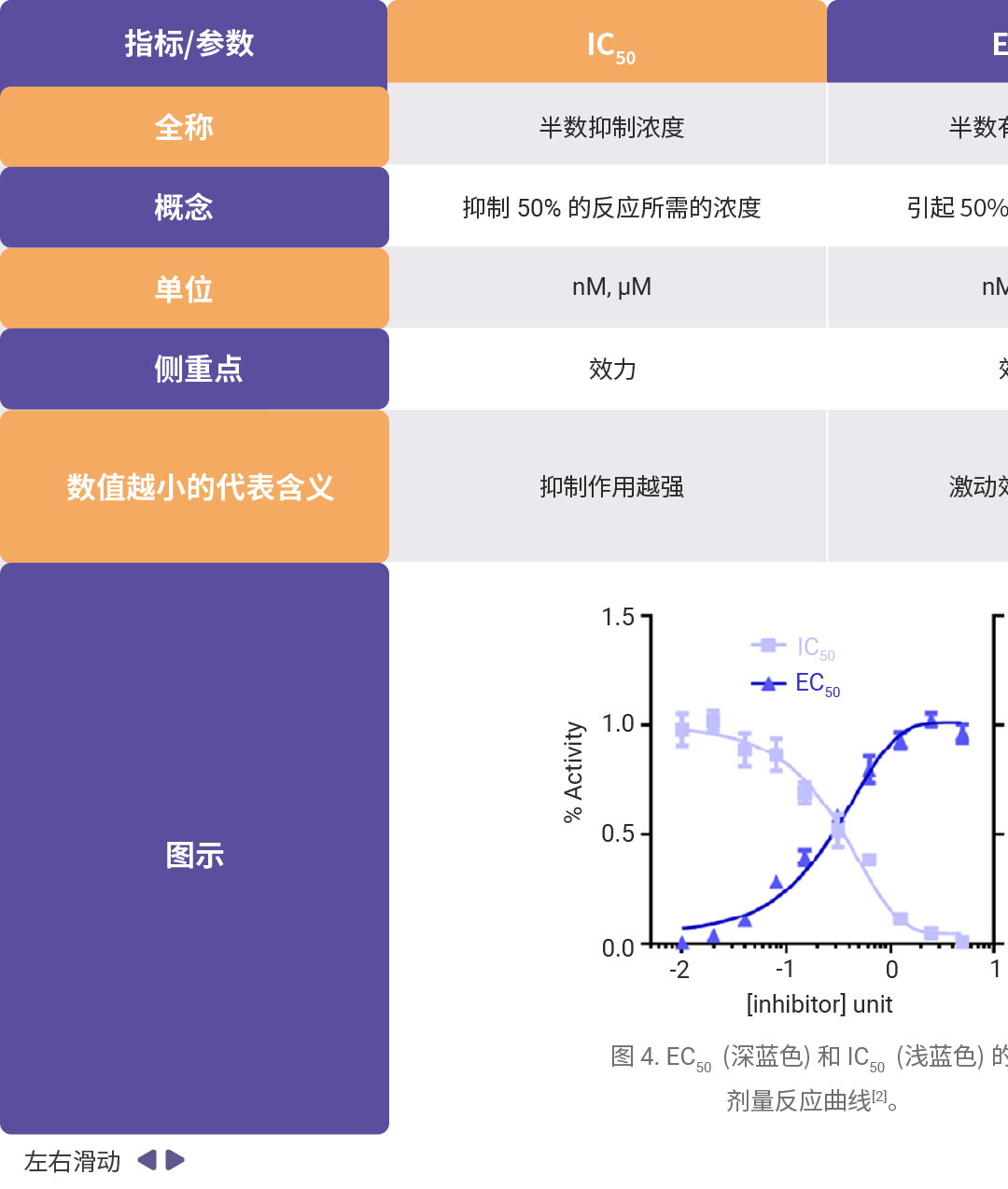
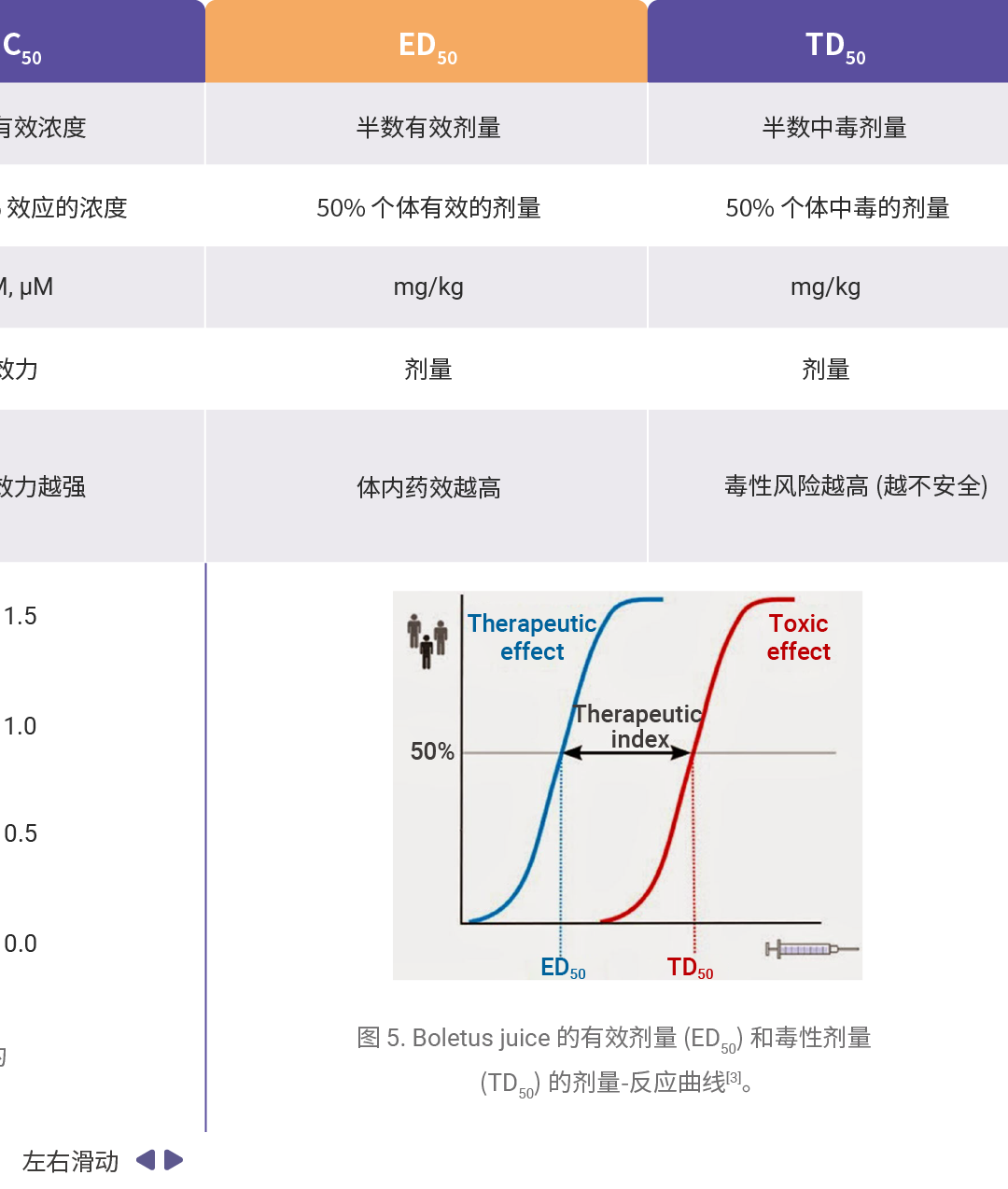
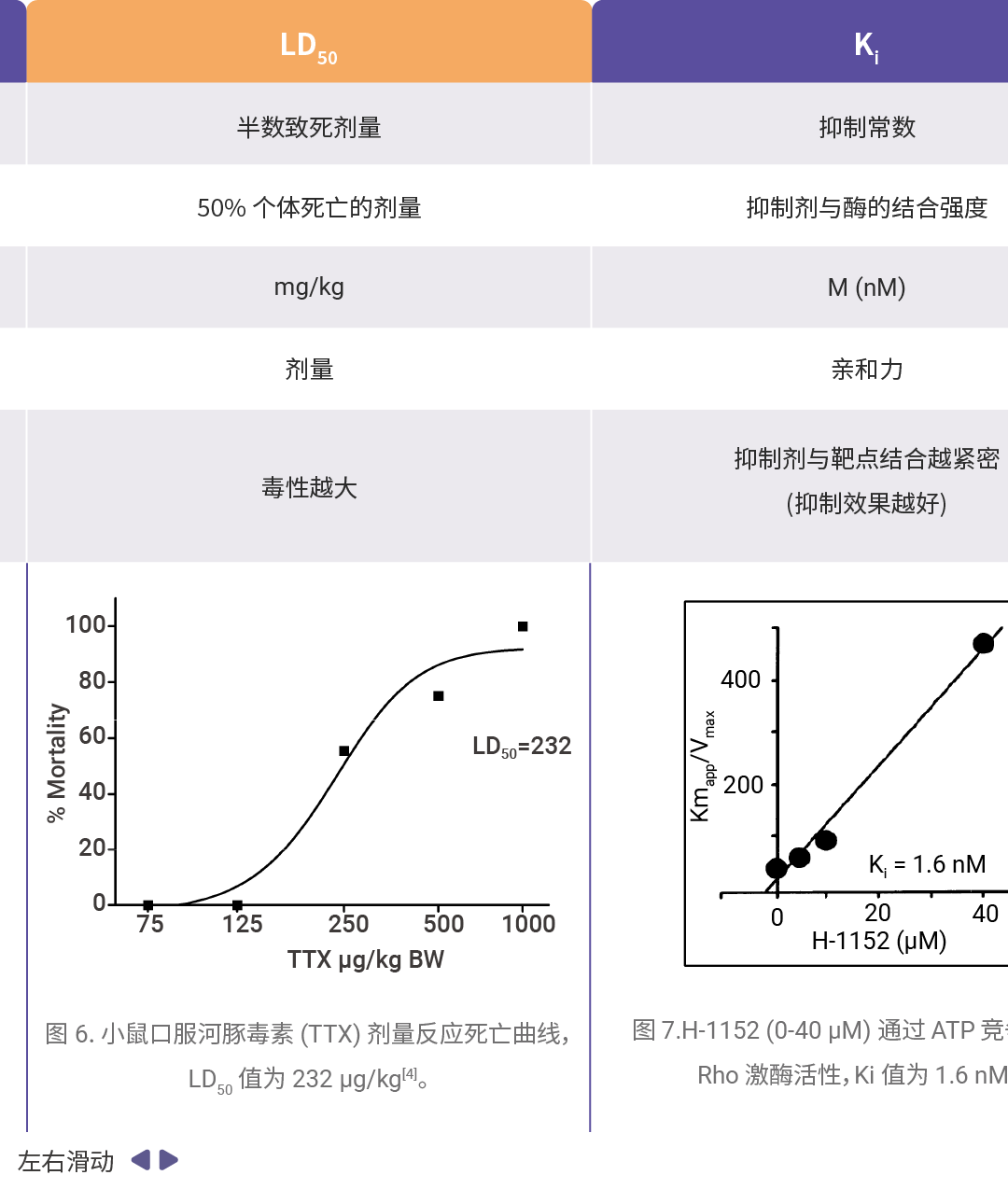
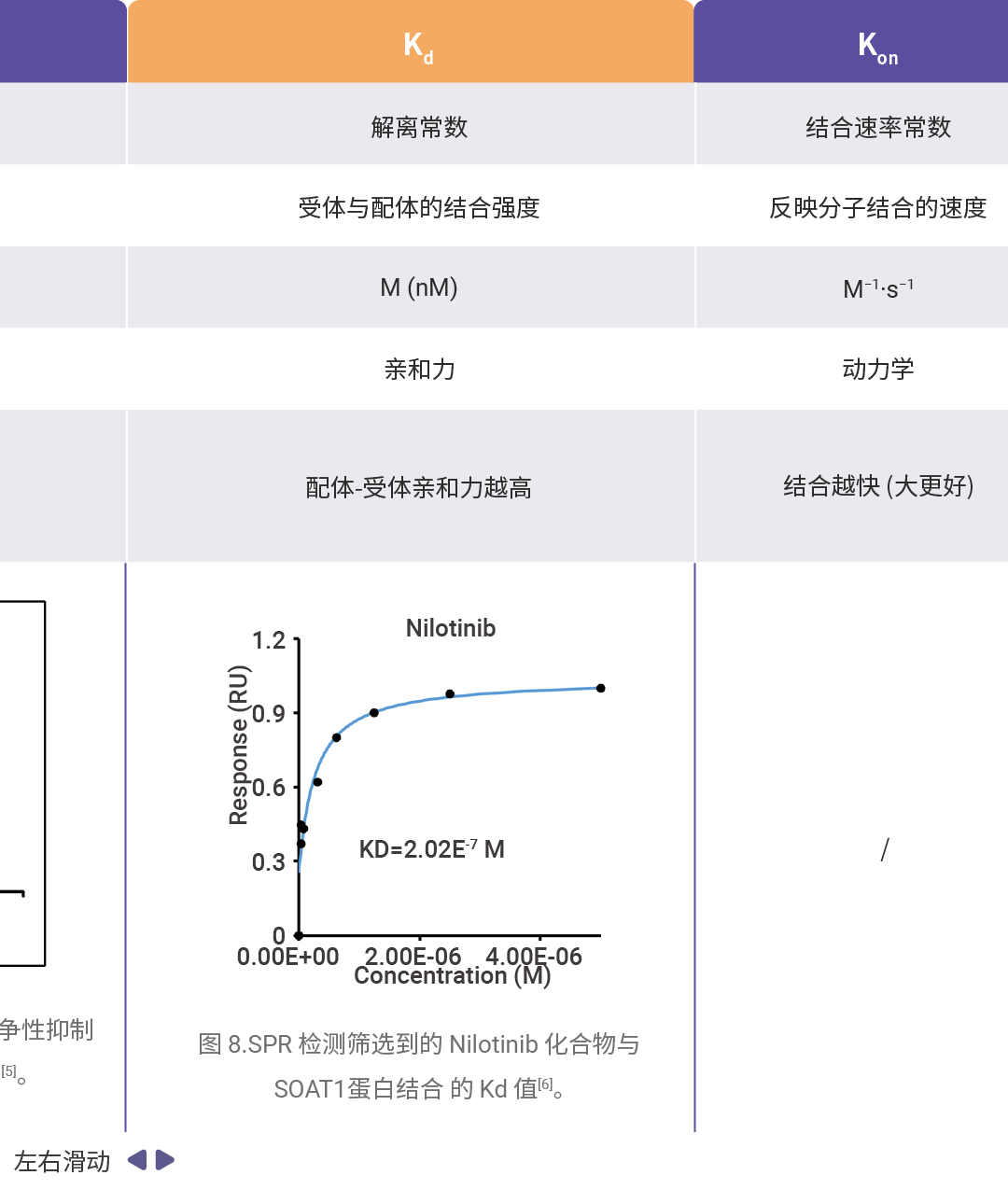
药效学和毒理学主要评价参数可详见往期:一图搞懂!IC₅₀、EC₅₀、ED₅₀......,汇总如下表所示。

Section.03
抗菌药物的 PK/PD
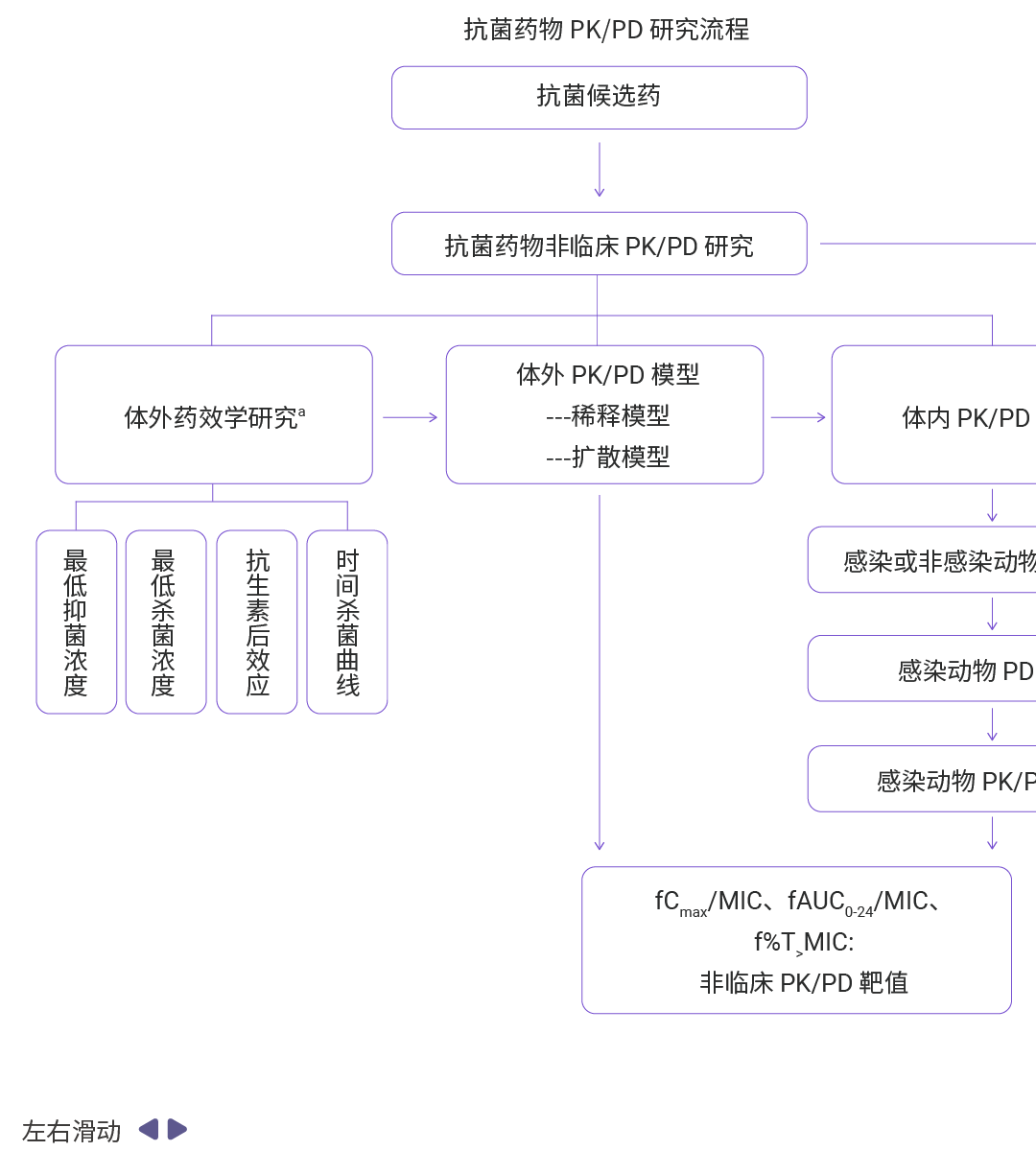
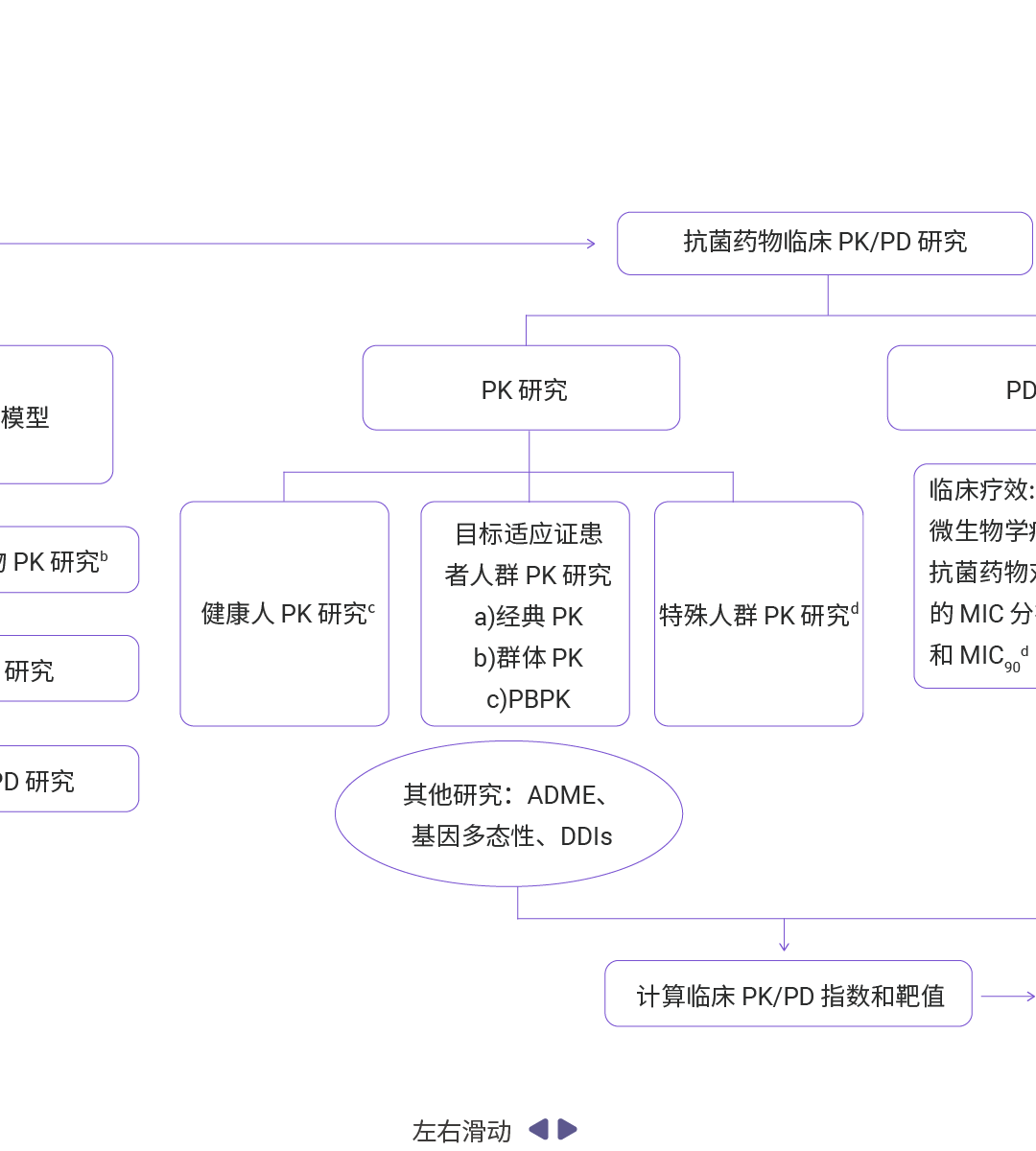
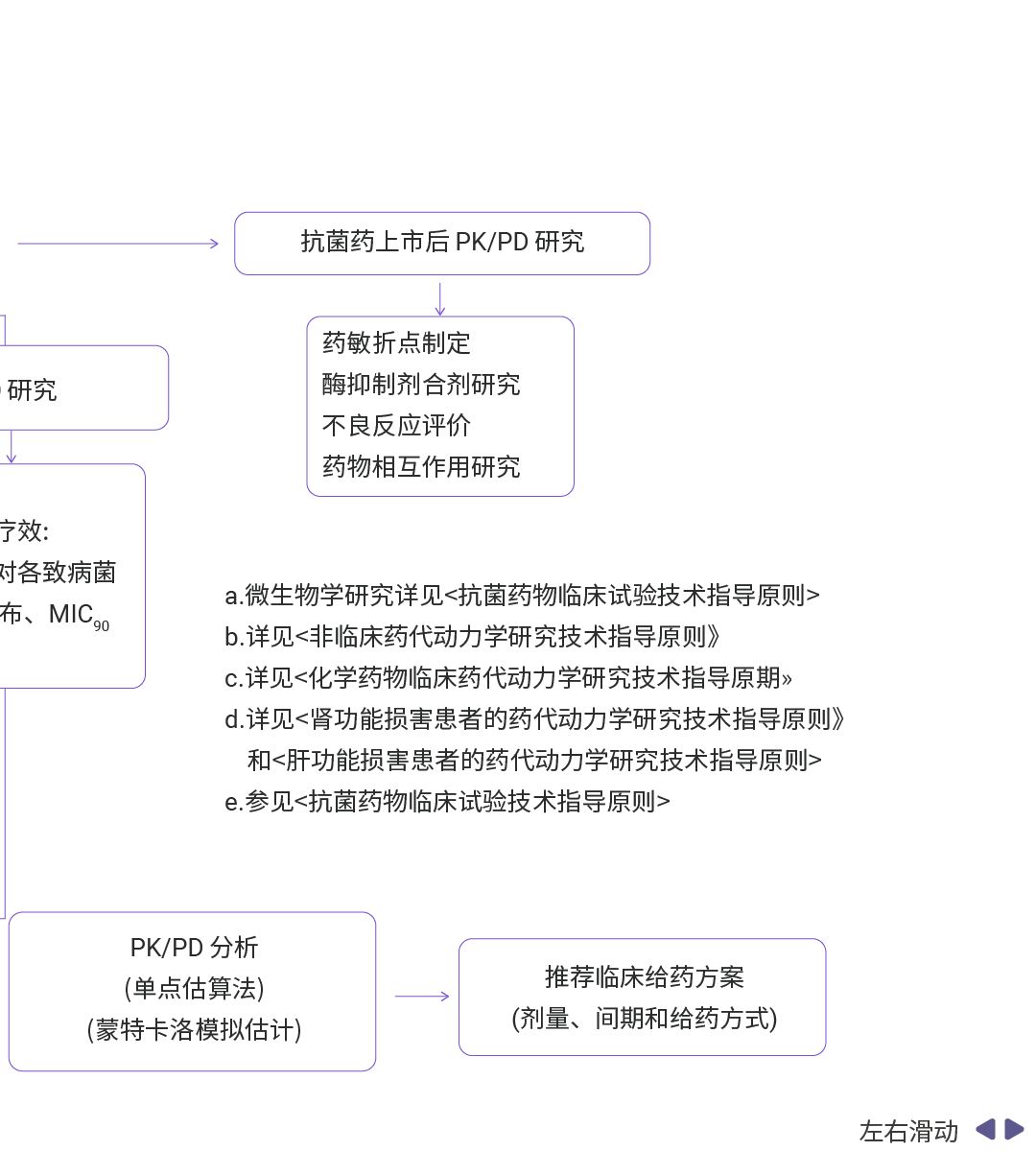
图 9. 抗菌药物 PK/PD 研究流程概览[7]。
抗菌药物 PK/PD 研究的基础参数除了 AUC、Cmax、t1/2 外,还包括 MIC、PAE、Cmax/MIC、AUC/MIC、T>MIC 等[7]。
MIC (最低抑菌浓度)
最低抑菌浓度,代表符号是 MIC,指在体外培养条件下,经过特定时间 (通常为 18-24 小时) 孵育后,能够完全抑制微生物 (细菌或真菌) 肉眼可见生长的最低药物浓度。
——MIC 越低,说明该药物抑制病原菌所需的剂量越小、抗菌活性越强。
-
通过测定标准菌株或临床分离株的 MIC,可判断菌株是否产生耐药。如果特定抗生素的 MIC 值异常偏高,通常意味着该细菌已产生抗药性。
-
概念区别:最低杀菌浓度,代表符号是 MBC,指能杀死 99.9% 以上供试微生物的最低药物浓度,通常 MBC ≥ MIC。
PAE (抗生素后效应)
抗生素后效应,代表符号是 PAE,指细菌短暂接触抗菌药物后,即使血药浓度降至最低抑菌浓度 (MIC) 以下或被完全清除,其生长仍受到持续抑制的现象。
也就是说,药物虽然已经消失,但细菌仍需经过一段时间的"复苏期"才能恢复正常的生长和分裂。在此效应期间,受损的细菌更容易被机体的白细胞识别和吞噬。利用 PAE 特性,临床可以实施更长间隔的大剂量给药 (如每日一次) ,减少毒副作用并提高疗效。
Cmax/MIC
一般将抗菌药物分为浓度依赖性药物和时间依赖性药物两大类。
Cmax/MIC,峰浓度与最低抑菌浓度的比值,是评估浓度依赖性抗菌药物 (如氨基糖苷类、喹诺酮类和硝基咪唑类) 疗效的核心 PK/PD 参数。
——该比值越大,说明药物对细菌的杀灭能力越强,通常 Cmax/MIC ≥ 8~10。当比值达到此标准时,药物的杀菌效果最显著。
AUC/MIC
AUC/MIC,指血药浓度-时间曲线下面积 (AUC) 与最低抑菌浓度 (MIC) 的比值。
——AUC/MIC 比值越高,说明药物在体内的浓度高于细菌致死或抑制浓度的持续时间和程度越充足,代表杀菌效果越好。
浓度依赖型抗菌药物的杀菌效果主要取决于药物浓度。通常要求 AUC/MIC 达到一定数值 (如 ≥30-125) 才能保证疗效并防止耐药。
时间依赖型抗菌药物 (如糖肽类,如万古霉素) : 对于重症感染,临床常以特定时间段的 AUC 与 MIC 的比值 (例如 AUC/MIC≥400) 作为万古霉素等药物的监测靶目标,以确保用药既有效又安全。
T>MIC
游离药物浓度在对病原菌 MIC 的 4~8 倍内,杀菌效果与浓度相关;但超过该浓度范围后,杀菌速率达饱和状态,其杀菌效果与药物浓度超过病原菌 MIC 时间的长短有关。
T>MIC,指在两次给药的间隔时间内,体内的实际抗生素浓度保持在 MIC 之上的持续时间,占给药间期的百分比,通常要求达到给药间隔的 40%~50% 以上。
产品推荐
Thiazolyl Blue (HY-15924)
www.medchemexpress.cn/MTT.html
MTT 是一种黄色的水溶性四唑盐,活细胞线粒体中的脱氢酶可以将 MTT 还原成不溶性的紫色结晶甲瓒,通过测量甲瓒的颜色变化来反映细胞活力。
CCK8 (HY-K0301)
www.medchemexpress.cn/inhibitor-kit/cell-counting-kit-8.html
WST-8 在电子载体 1-Methoxy PMS 的作用下,可以被细胞内脱氢酶还原成水溶性的橙黄色甲臜 (formazan)。
WST-1 (HY-136976)
www.medchemexpress.cn/wst-1.html
WST-1 诱导细胞内线粒体脱氢酶进行 NADH 依赖的酶切反应,释放出水溶性的甲臜产物,通过测定 450 nm 处的吸光值来反映细胞活力。
参考文献
[1]化学药物非临床药代动力学研究技术指导原则
[2]Zaman A, et al. Quantitative Framework for Bench-to-Bedside Cancer Research. Cancers (Basel). 2022 Oct 26;14(21):5254.
[3]Gui, H., et al. Effects of Boletus Poisoning on Estrogen Receptors and Neurotransmitters in Rats Based on ERk1/2 Pathway. Neural Process Lett 55, 193–203 (2023).
[4]Abal P, et al. Acute Oral Toxicity of Tetrodotoxin in Mice: Determination of Lethal Dose 50 (LD50) and No Observed Adverse Effect Level (NOAEL). Toxins (Basel). 2017 Feb 24;9(3):75.
[5]Ikenoya M, et al. Y. Inhibition of rho-kinase-induced myristoylated alanine-rich C kinase substrate (MARCKS) phosphorylation in human neuronal cells by H-1152, a novel and specific Rho-kinase inhibitor. J Neurochem. 2002 Apr;81(1):9-16.
[6]Wang Z.H, et al. High-afnity SOAT1 ligands remodeled cholesterol metabolism program to inhibit tumor growth. BMC Medicine. 2022 Aug 9;20(1):292.
[7]抗菌药物药代动力学/药效学研究技术指导原则
