- 移动端


MedChemExpress LLC品牌商
14 年
手机商铺
商家活跃:
产品热度:
- NaN
- 0
- 0
- 2
- 2

JQ1抑制剂-MCE(MedChemExpress)
询价
推荐产品




公司新闻/正文
Nature 发文 | MASH 致癌机制又有新发现!_ MedChemExpress(MCE 中国)
212 人阅读发布时间:2025-06-10 15:57

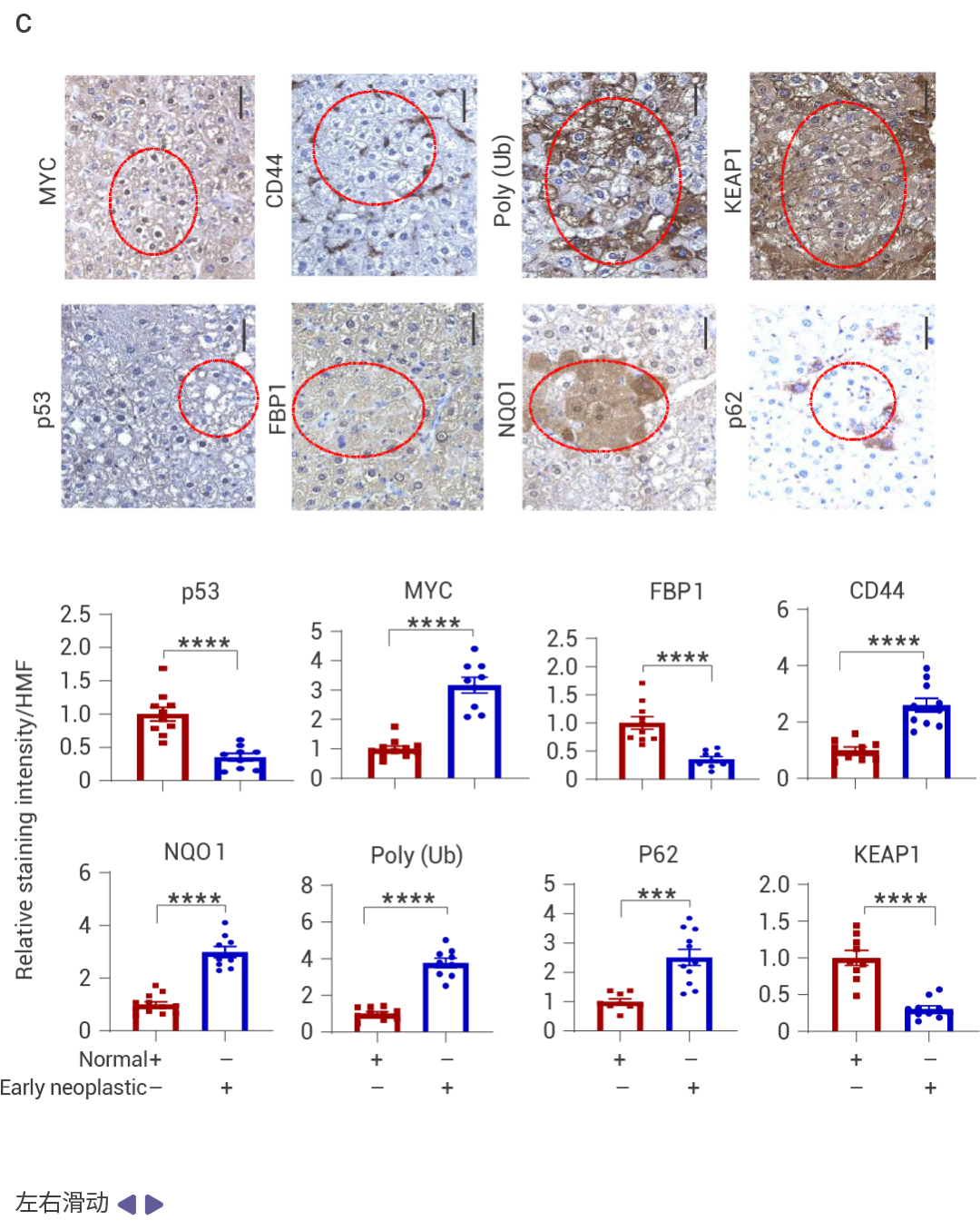
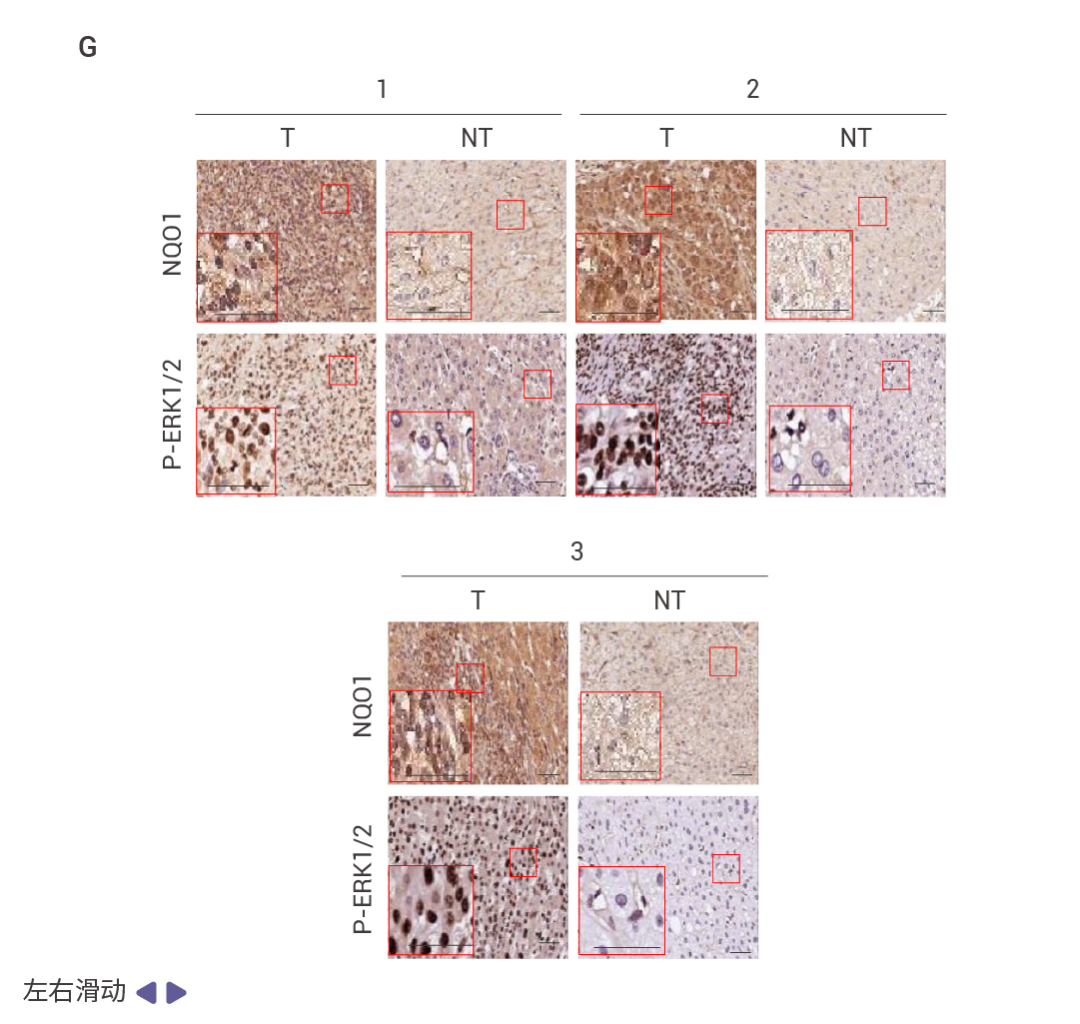
人类 HCC 中的 p53 和 FBP1 表达下降
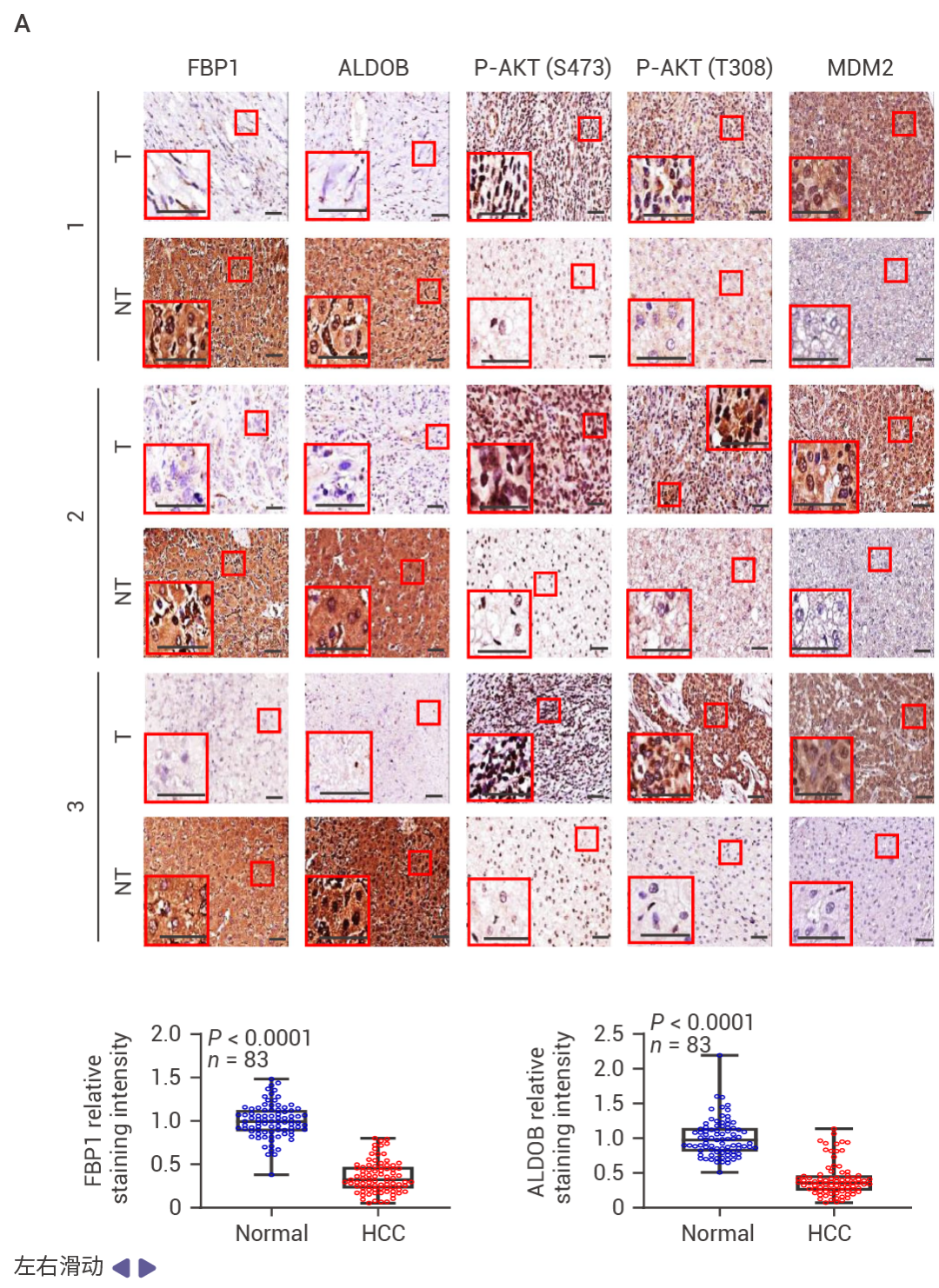
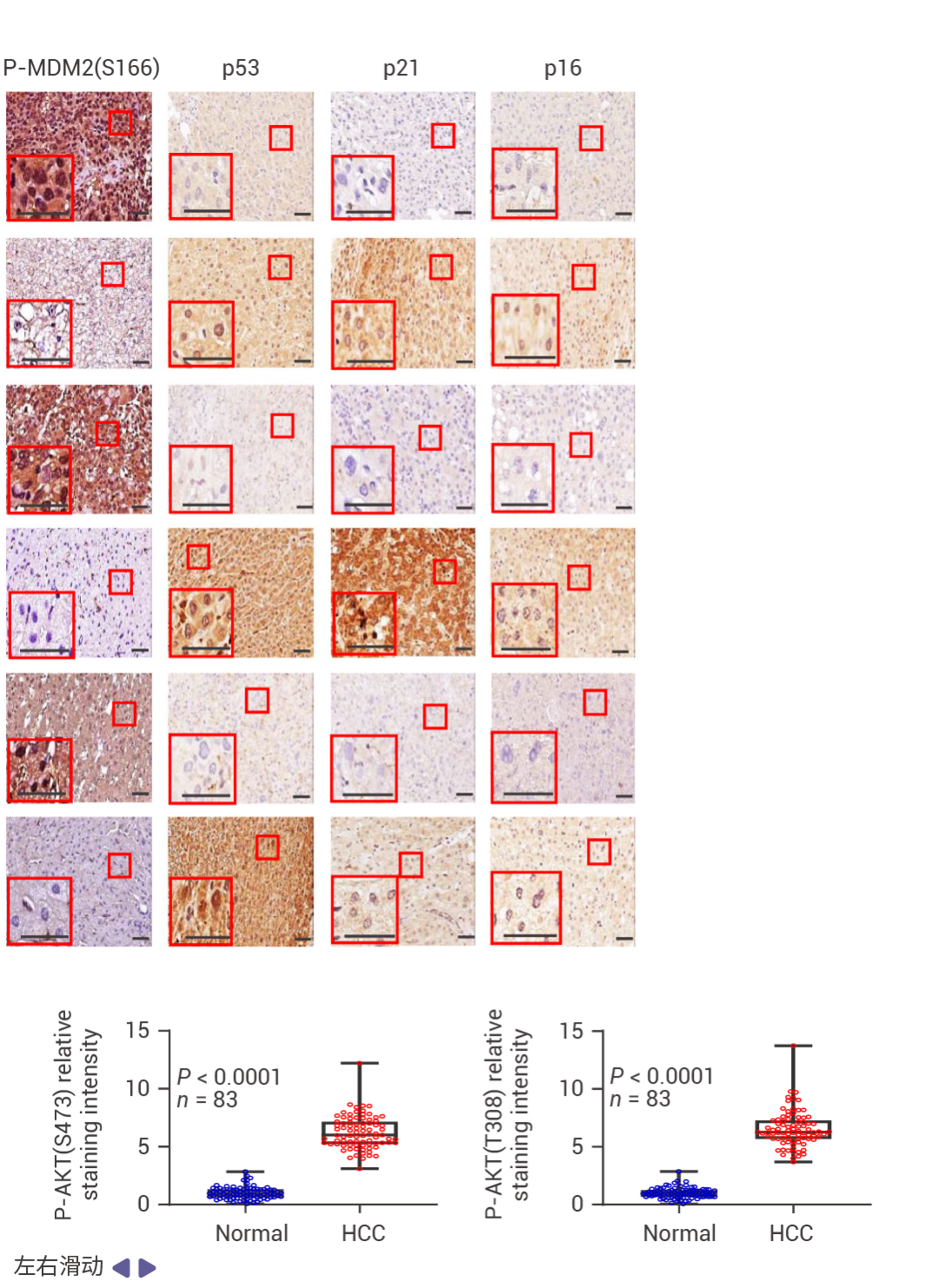
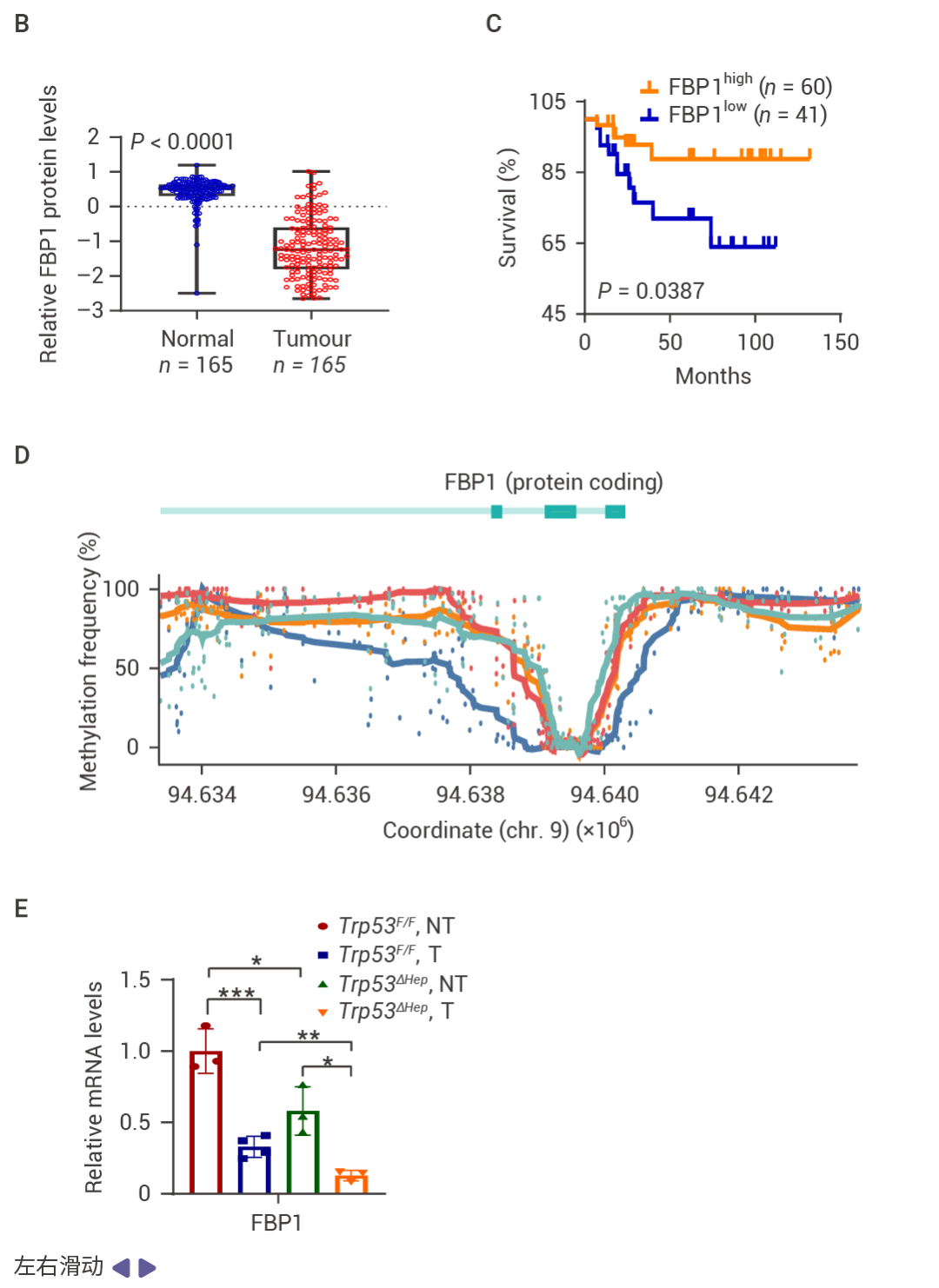
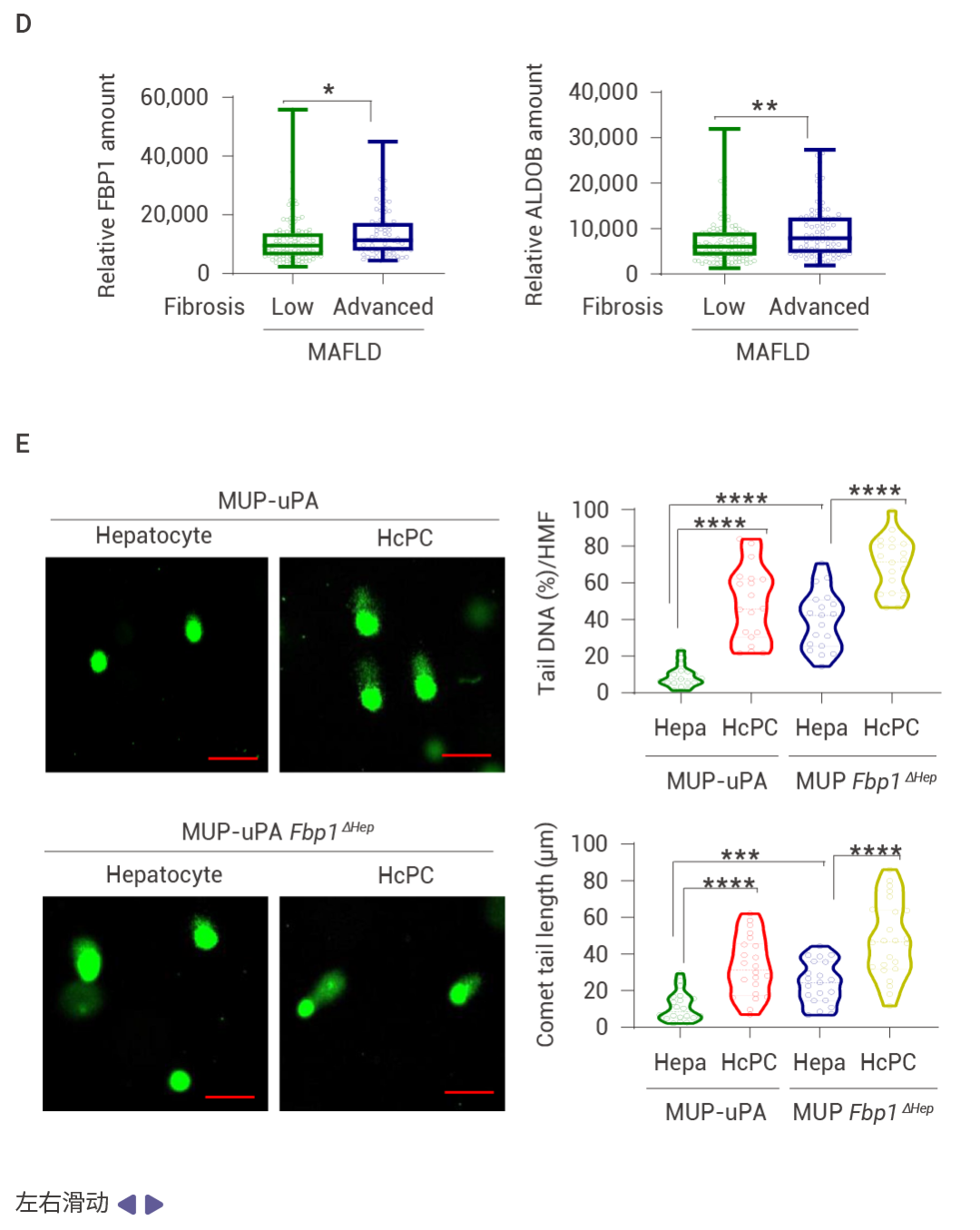
MASH 提高的 FBP1 和 p53 抑制 HCC
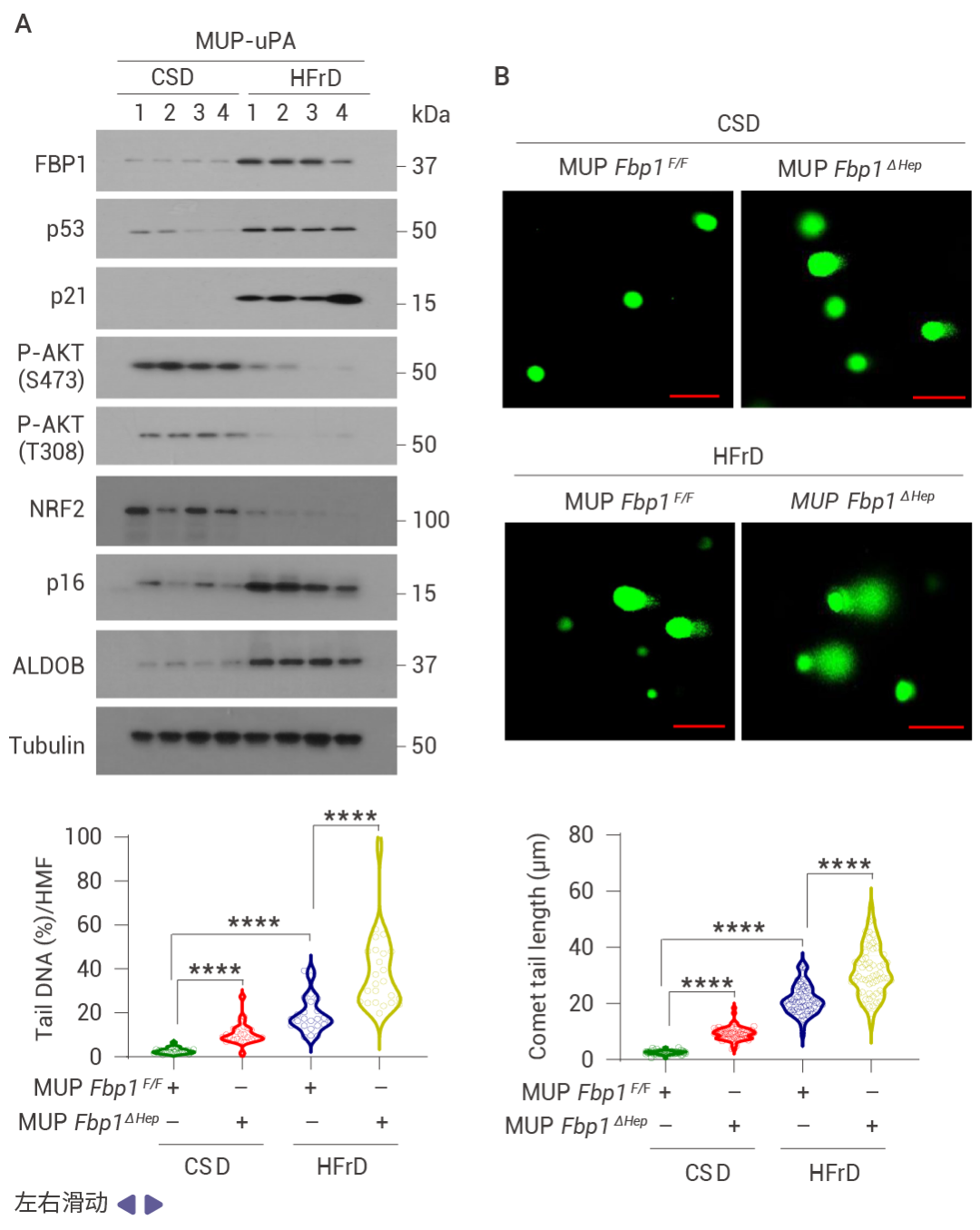
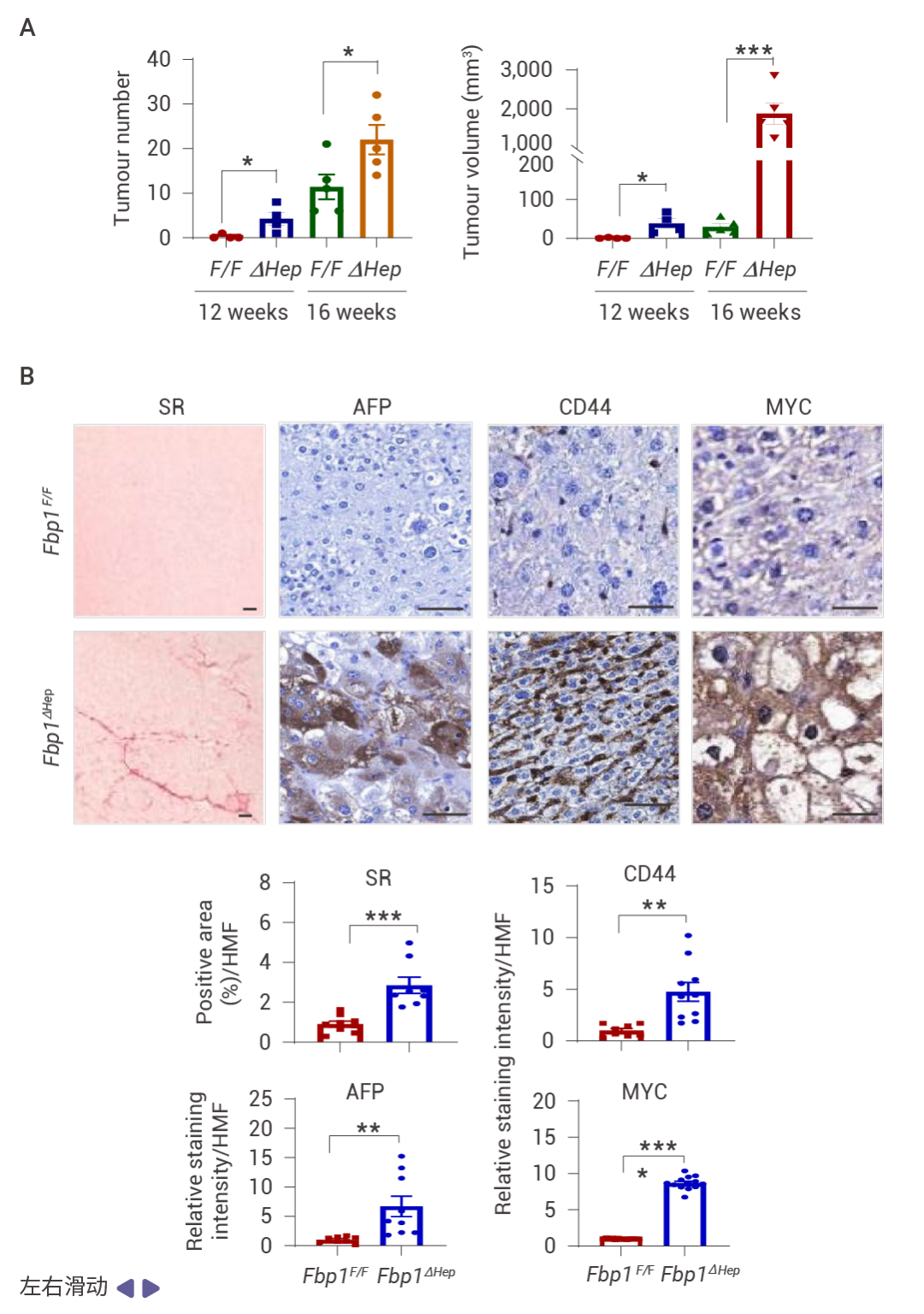
FBP1 的缺失通过 AKT 激活促进肿瘤发生
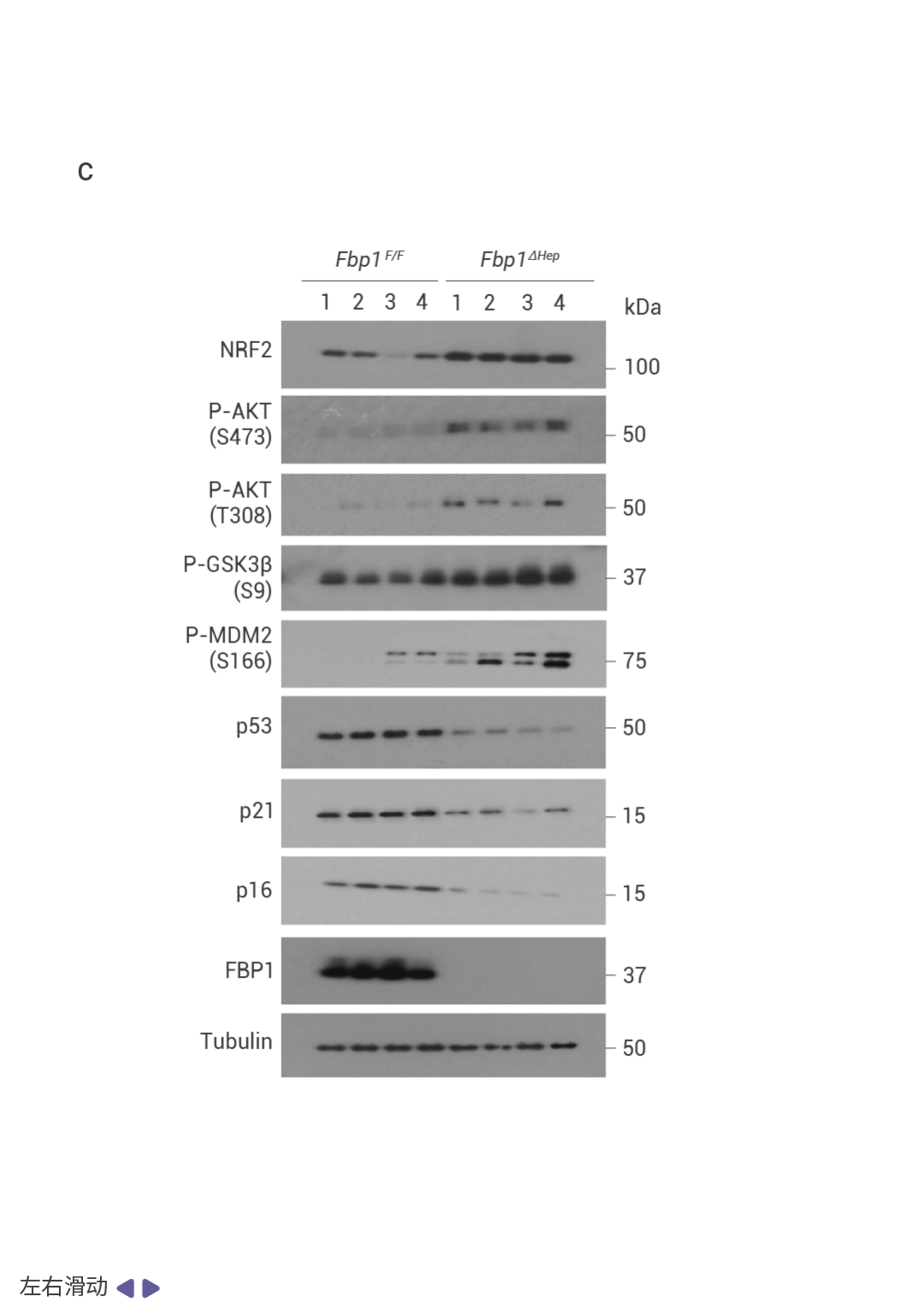
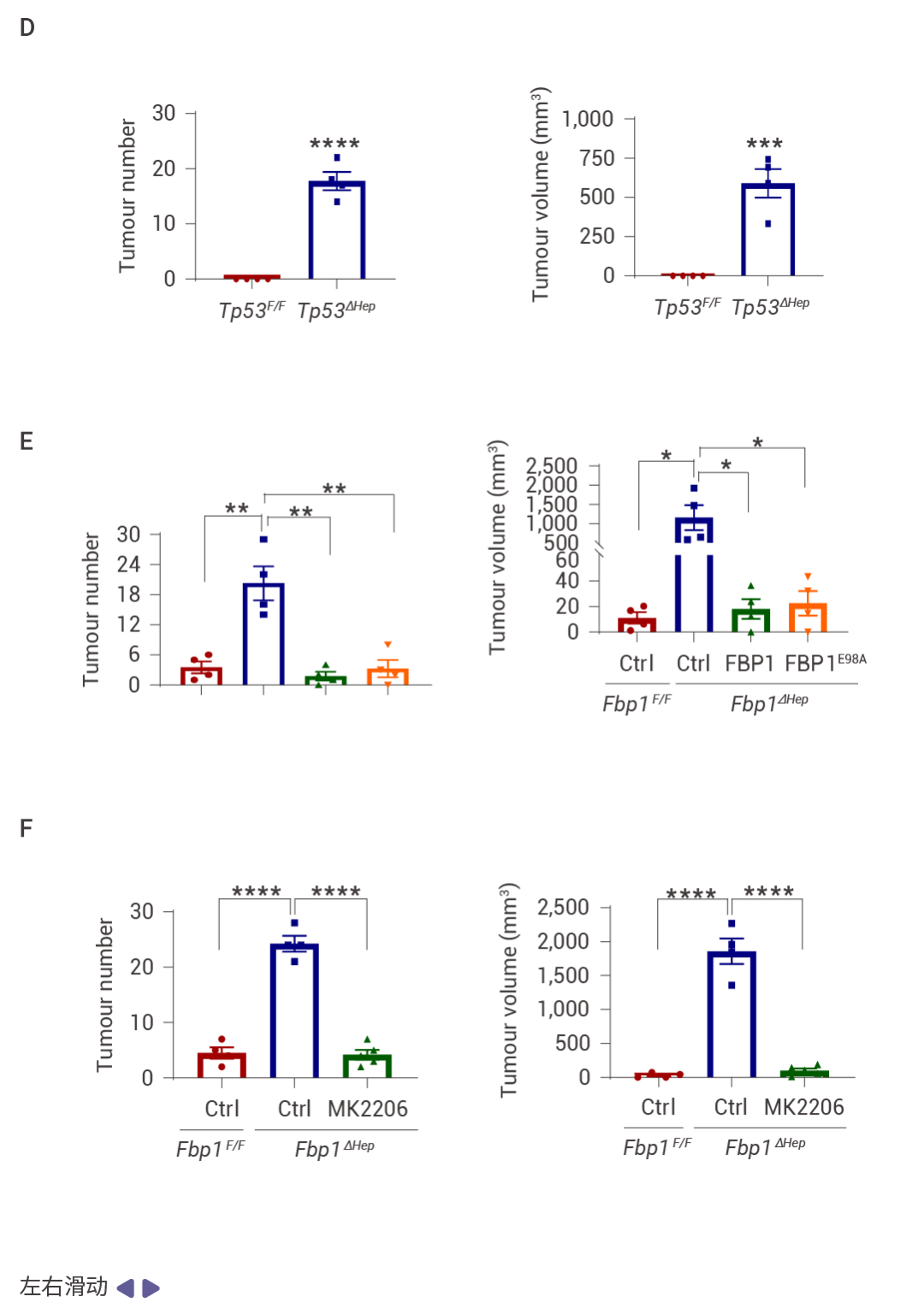
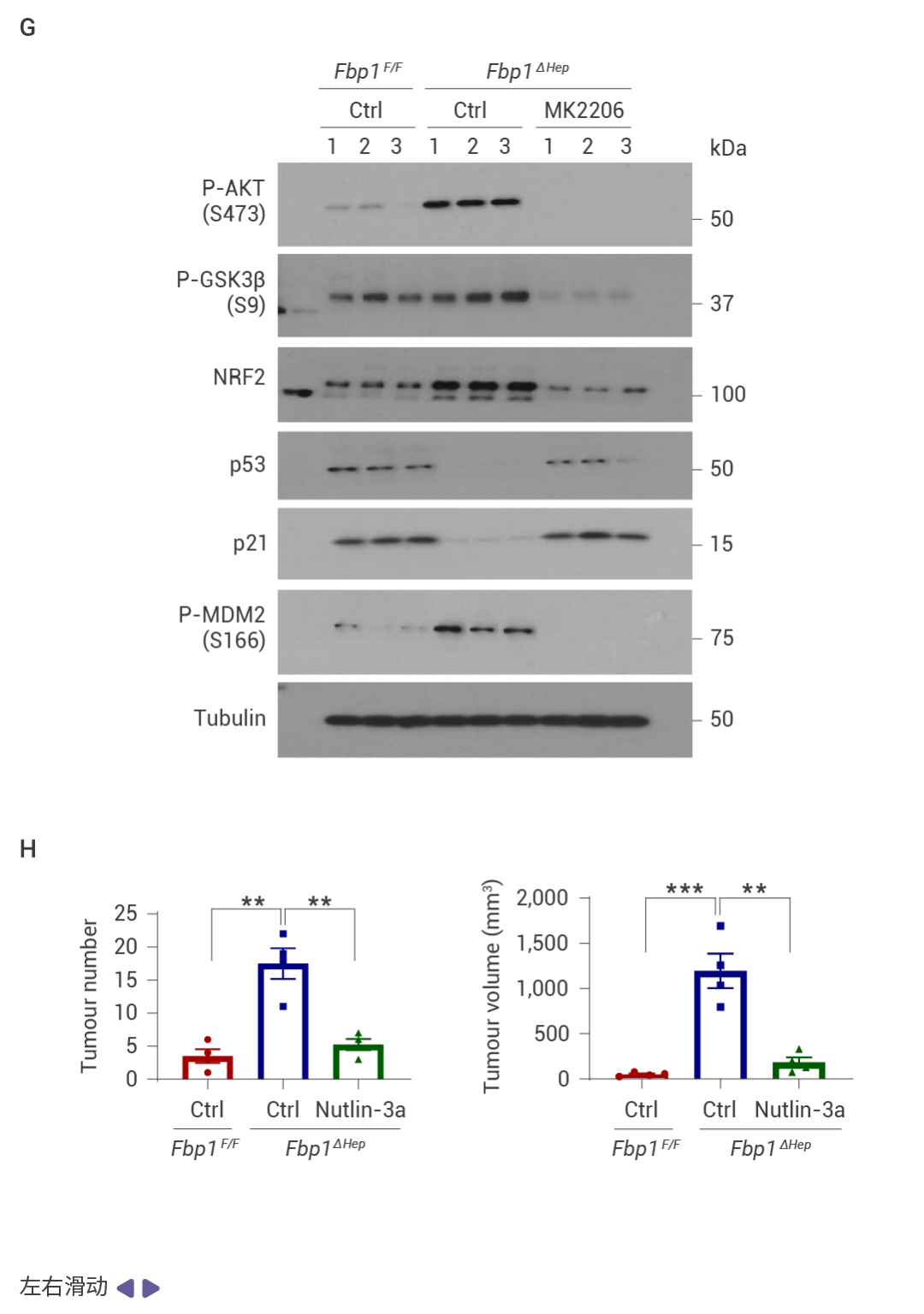
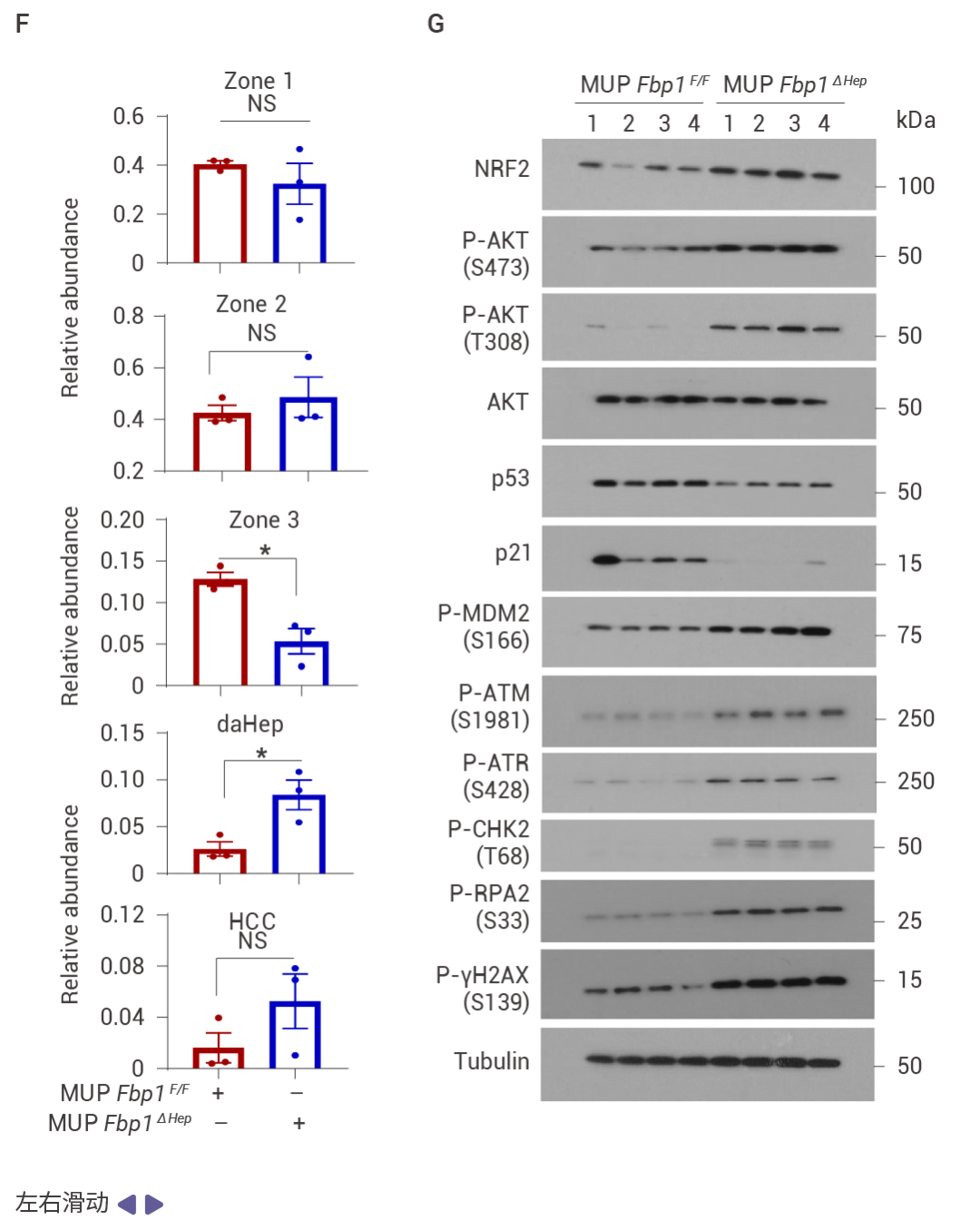
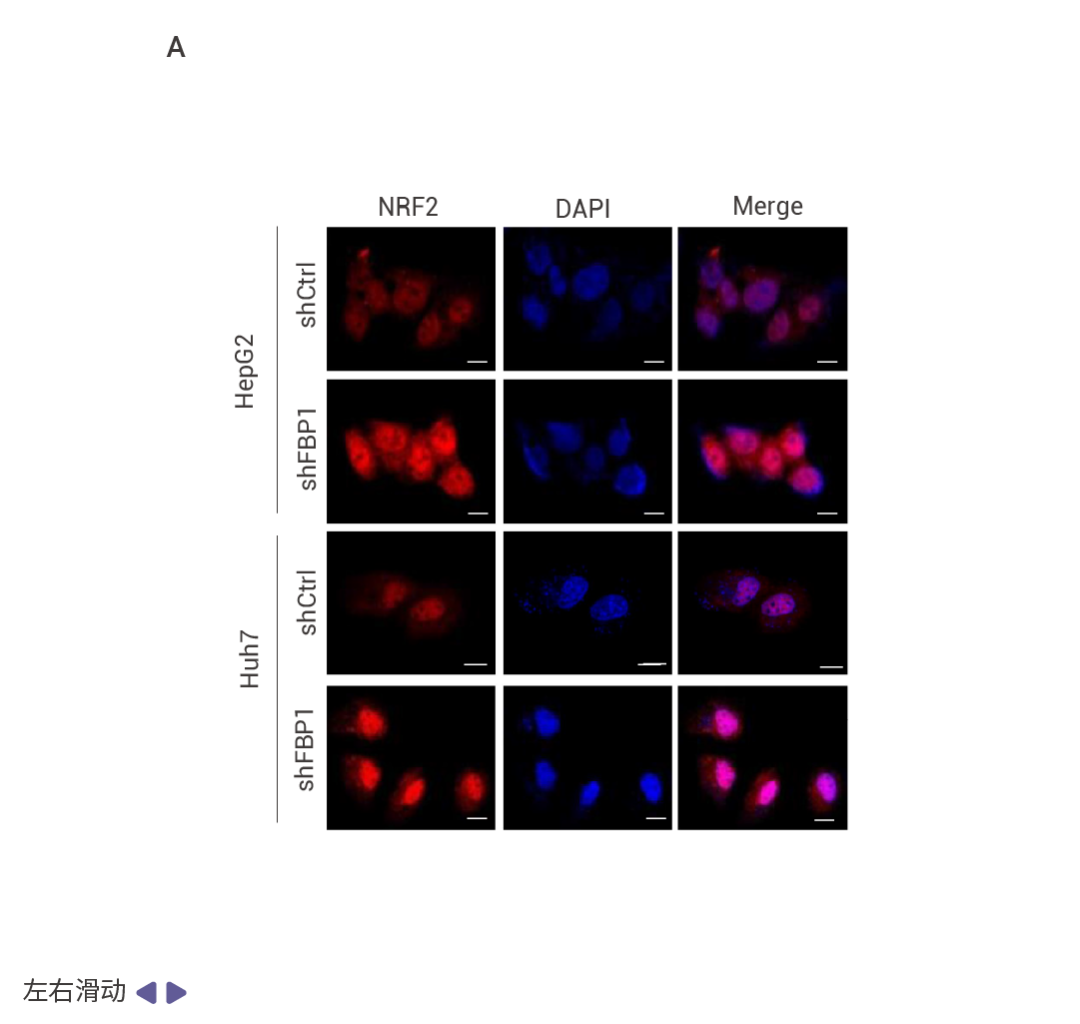
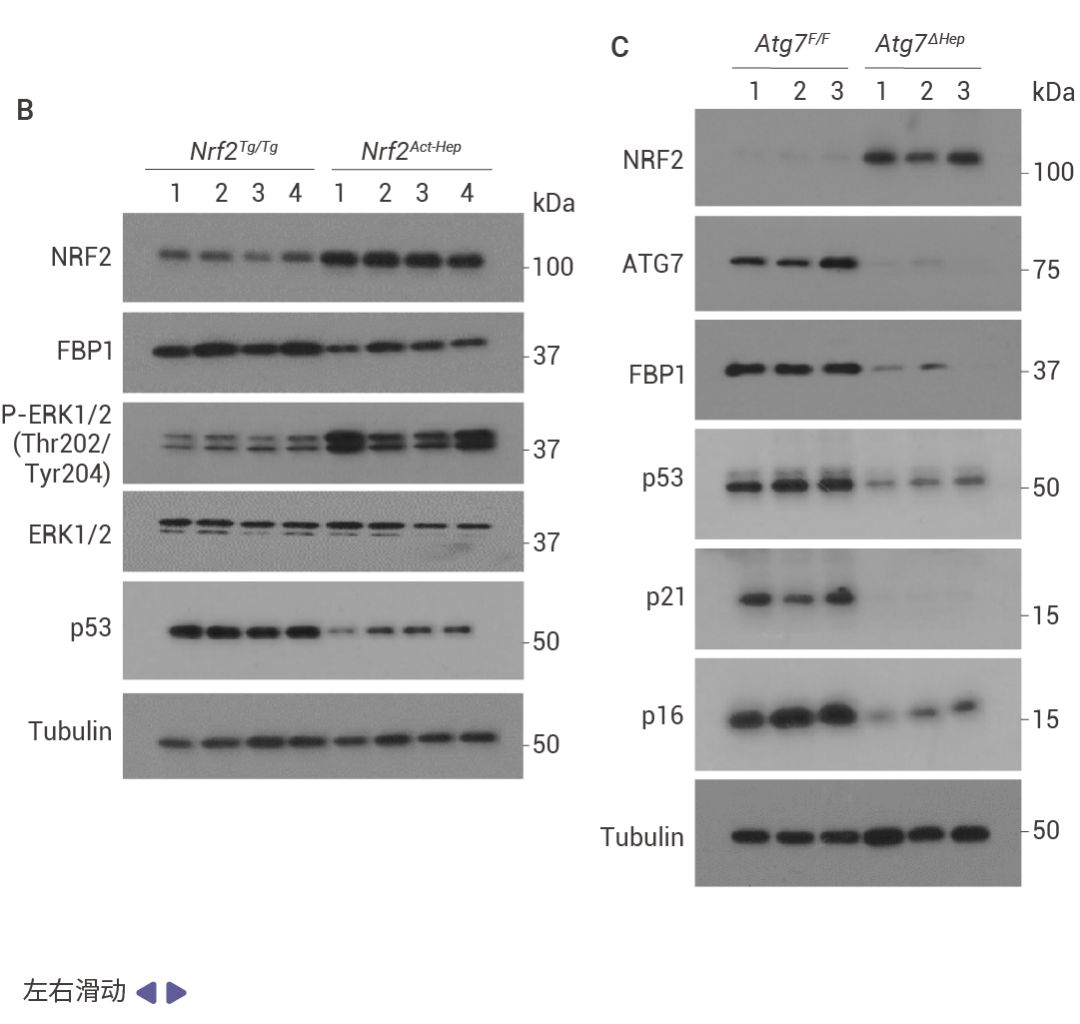
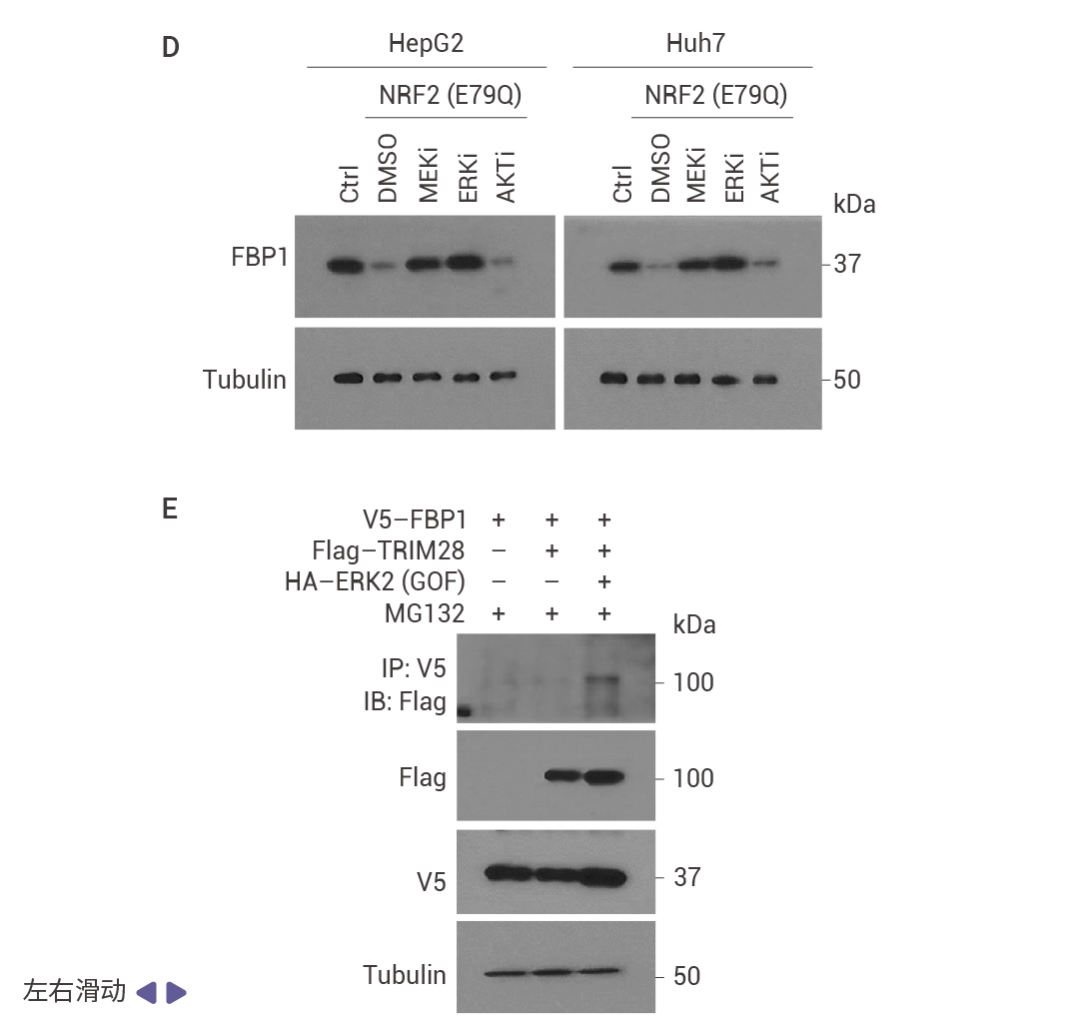
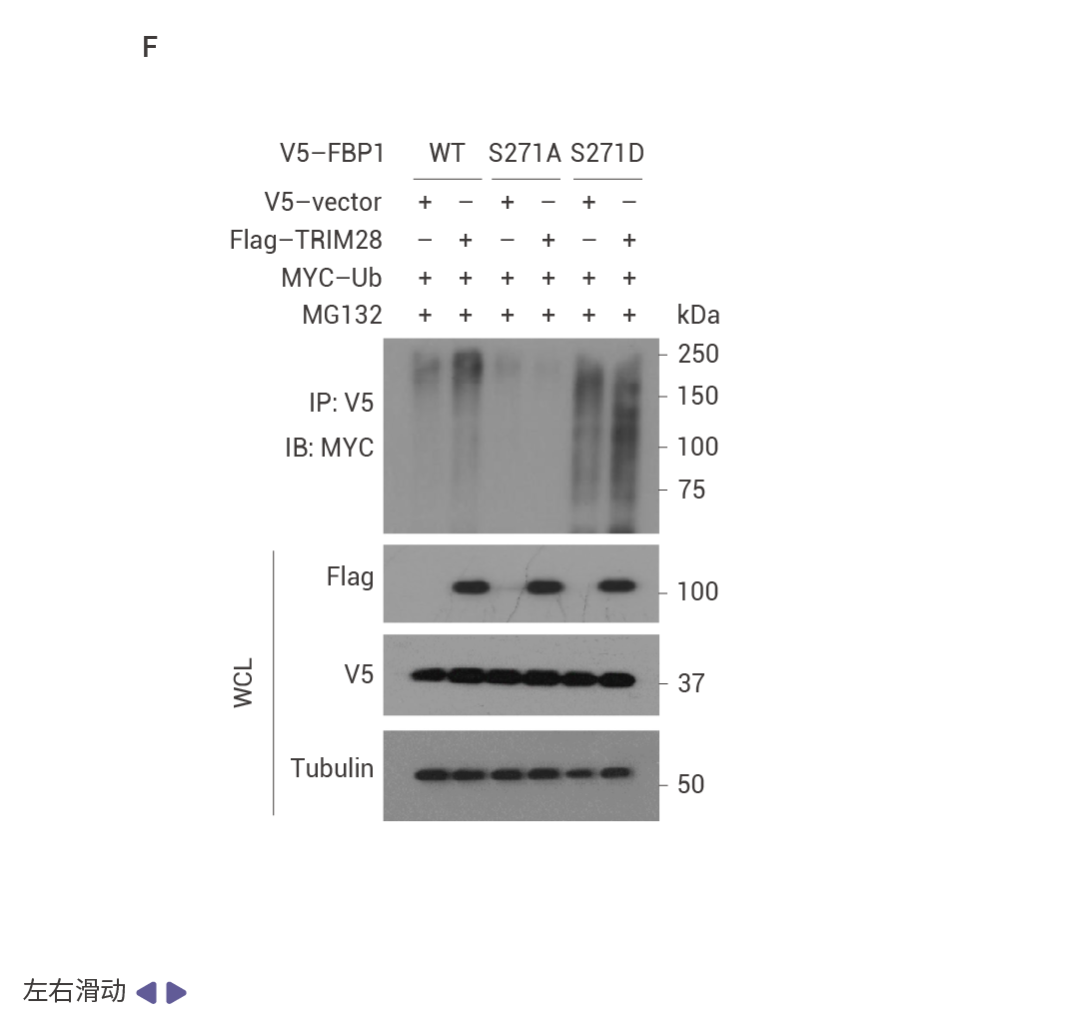
相互调控的 NRF2–FBP1 交互作用
|
|
|
|
|
|
|
|
|
|


